
EMA(European Medicines Agency:欧州医薬品庁)及びFDA(Food and Drug Administration:アメリカ食品医薬品局)では、既にニトロソアミン類の管理が厳格化されており、今後は日本においても同様の厳格管理が求められることが予想されています。
このような中でシオノギファーマは、データインテグリティの観点でもGMPの要求事項を満たす高感度分析法を確立しました。本稿では規制当局の動向ならびに高感度分析法の開発について概説します。
EMAとFDAのニトロソアミン類の規制の動向
EMAは、2019年9月にジェネリック医薬品やOTC製品を含むすべての医薬品の製造販売業者に対し、ニトロソアミンの混入リスクを評価し、適切なリスク低減策を講じるよう通達しました。その後、FDAも医薬品中のニトロソアミン類の管理に関するガイダンスを2020年9月に発出しました(末尾の参考情報を御参照ください)。
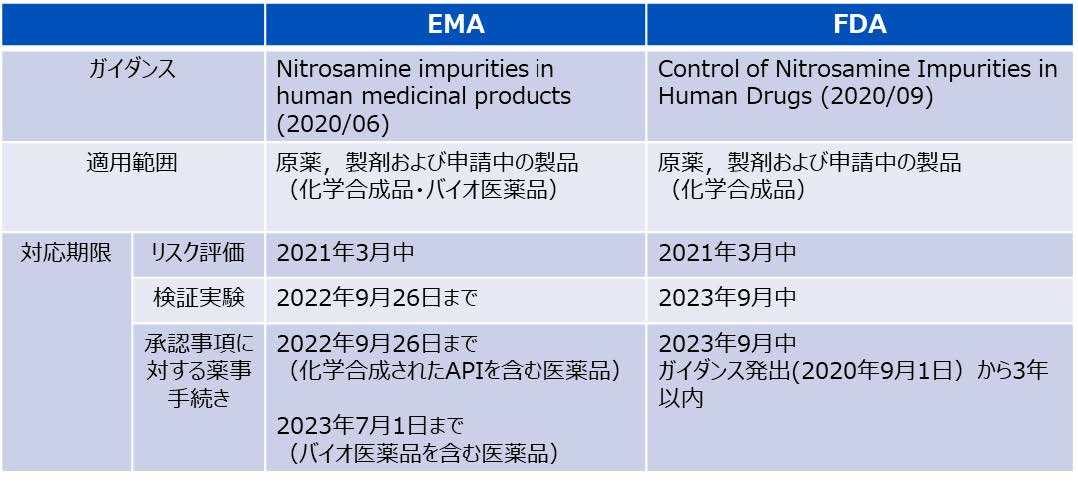
医薬品に含まれているニトロソアミンのリスク評価を実施し、適切なリスク低減策を講じる手順に関しては、EMAと米国FDAが発出したガイダンスに大きな差はありません(下表参照:【適用範囲と対応期限の留意点】)。唯一の大きな違いは、EMAガイダンスでは適用範囲に“バイオ医薬品”が含まれていることです。
【適用範囲と対応期限の留意点】
リスク評価から変更管理までのプロセス
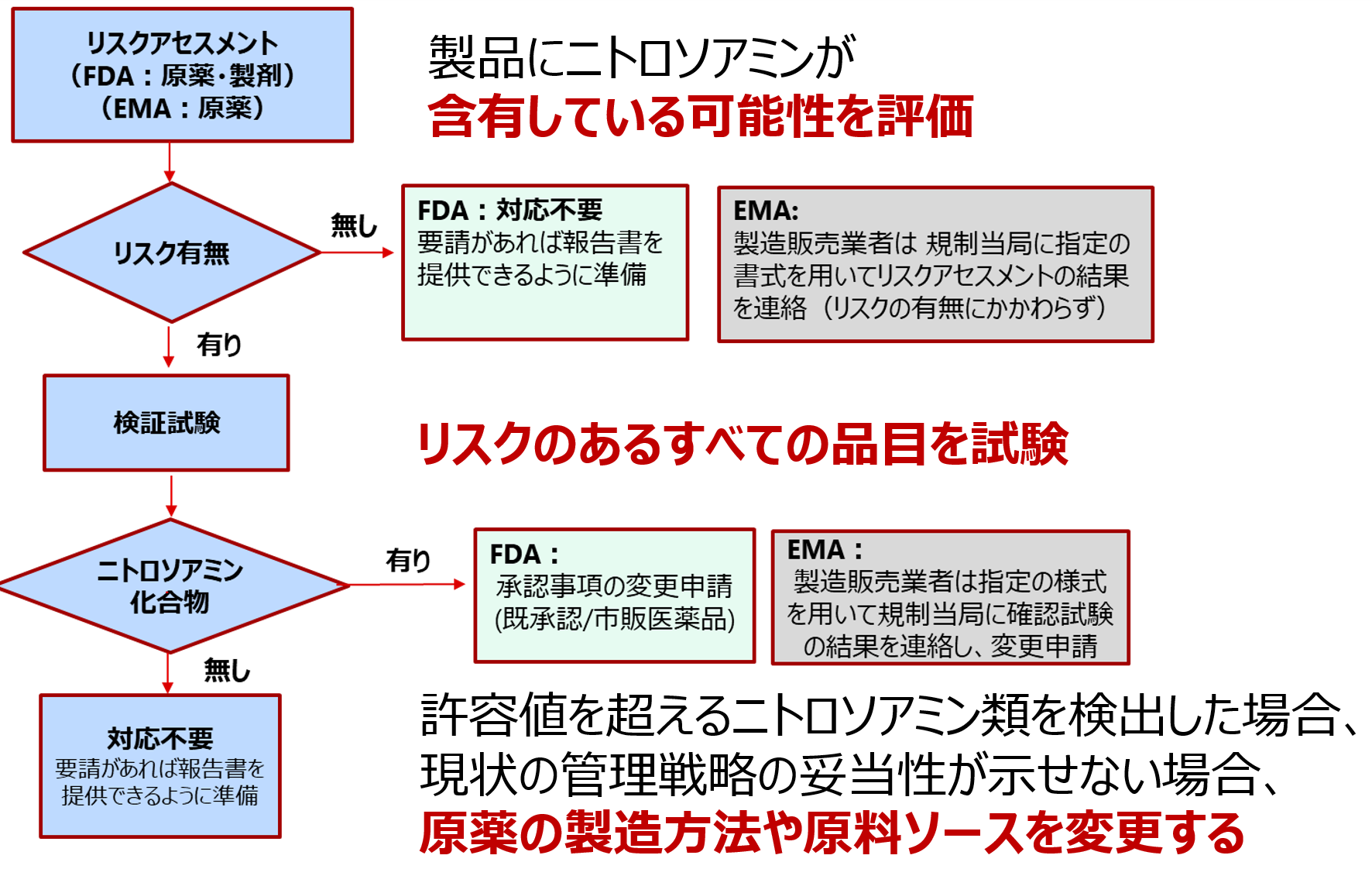
EMAもFDAも製造販売業者が実施すべきリスク評価から変更管理までのプロセスは大きく分けて3つのステップ(①リスクアセスメント,②検証試験,③製法や原料ソースの変更管理)で構成されます。
まず始めのステップとして、原薬製造業者はニトロソアミン不純物の可能性を特定するため、原薬の製造工程を照査しリスクアセスメントを実施します。ニトロソアミン不純物の混入の可能性がないと判断された場合は、それ以上の措置を講じる必要はありません。
一方で、混入のリスクが否定できない場合には、次のステップとして、適切な感度を有し、バリデートされた試験法を用いてバッチの検証試験を実施することが必要となります。実測の結果、許容値を超えるニトロソアミンが検出された場合には、根本原因を調査し、ニトロソアミン不純物を低減するために製造工程や原料ソースの変更が求められます。
高感度分析法の開発
ニトロソアミン類は、その著しい発がんリスクの可能性により、ppbオーダーの定量限界が要求されています。FDAのガイドラインは定量限界が0.03ppm以下の試験法を用いることを求めており、EMAにおいても検証試験には許容限度値の10%未満のレベルで混入の有無を評価できることを求めています。すなわち、規制の要求事項を満たすには高感度な分析法の設定が必要となります。
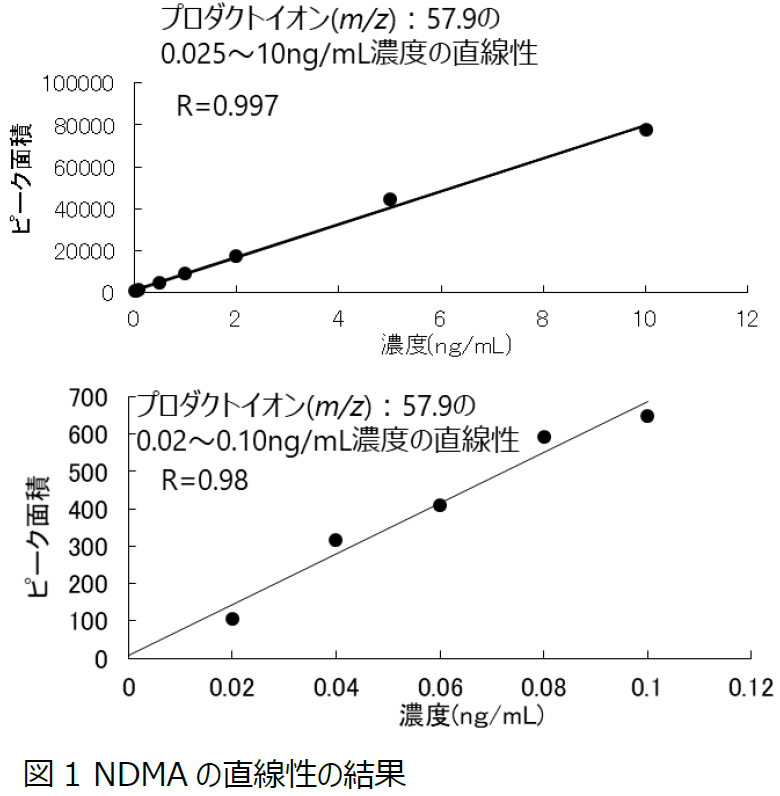
シオノギファーマではLC-MS/MSによる高感度分析法の検討を開始し、まずはNDMA(N-ニトロソジメチルアミン)水溶液の直線性および定量限界を求めました。プリカーサーイオン74.9に対して、プロダクトイオン42.9および57.9で比較したところ、プロダクトイオン57.9の方が、ベースラインノイズが少なく良好なクロマトグラムが得られたことから、プロダクトイオン57.9を定量に用いています。直線性の結果を図1に示します。0.025~10ppm*の範囲では相関係数0.997であり、低濃度側0.02~0.10ppm*でも相関係数0.98と良好な直線性を示しました。また、0.025ppm*濃度のS/N比は20以上であり、6回繰り返しの相対標準偏差は6.6%で、定量限界として十分な再現性を有していると判断しました(*1mg/mLの試料溶液に対して)。
ニトロソアミン類の分離条件
図2にシオノギファーマが開発した分析条件における、代表的な8種類のニトロソアミンのMSクロマトグラムを示します。DIPAとNMPAの溶出位置が近いですが、両者はMSにより識別可能で、特異性には全く問題ありません。8成分を同時に分析可能で、グラジエントの平衡化を含めて8分で分析が完了する迅速な条件を設定することができました。

NDMA: N-ニトロソジメチルアミン, NDEA: N-ニトロソジエチルアミン, NMBA: N-ニトロソ-N-メチル-4-アミノブタン酸, NEIPA: N-ニトロソエチルイソプロピルアミン, NDIPA: N-ニトロソジイソプロピルアミン, NMPA: N-ニトロソメチルフェニルアミン, MeNP: N-メチル-N‘-ニトロソピペラジン, NDBA: N-ニトロソジブチルアミン
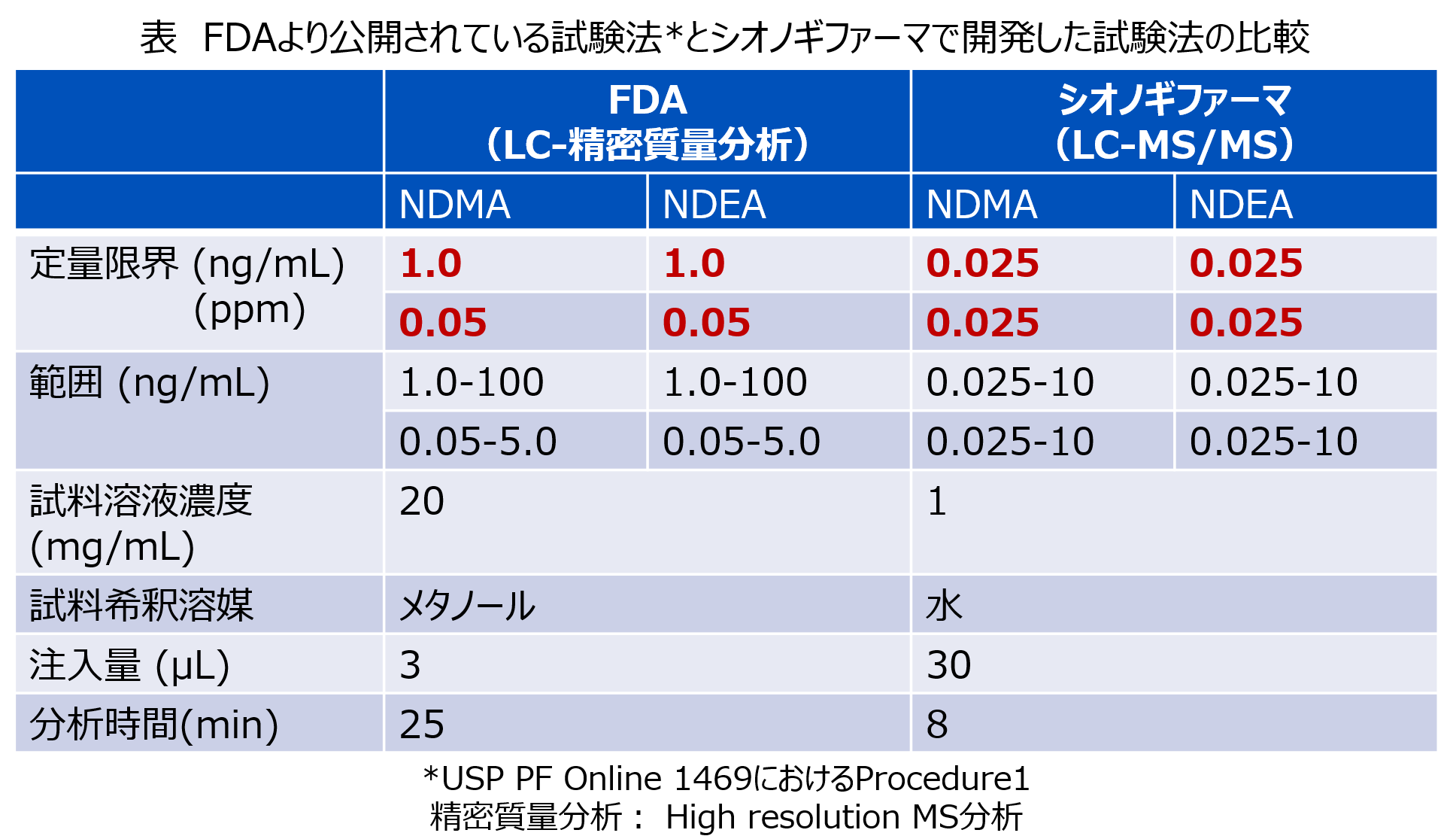
下表に、FDAにより公表されているロサルタン原薬および製剤中の6種のニトロソアミンのLC-精密質量分析とシオノギファーマが開発した分析条件を比較しています。既存のFDA試験法では、0.03ppm以下の定量限界を達成できておらず、より高感度な分析が必要となります。一方、シオノギファーマが設定した試験法では、定性・定量の面で優れたタンデム型の質量分析計を用い、また、試料溶媒を水とすることで注入量を増やすことが可能となり、定量限界0.025ppm、範囲 0.025~10ppmの測定が可能な方法を確立できました。また、Mass Lynx Securityソフトウェアを使用することによりデータインテグリティの観点においてもGMPの要求事項を満たすデータ取得が可能となりました。
以上、厳格化の流れにあるニトロソアミンの規制に対応する医薬品および原料の試験法として、十分な感度と正確性を有した分析法を確立できたと考えています。
まとめ
医薬品におけるニトロソアミン不純物の規制は、欧米においては化学合成された原薬を含む全ての製品に広がっており、今後、日本においても同様の管理が求められることが予想されます。プロセス評価のみでリスクを完全に否定できない場合には、製造販売業者は検証試験を実施する必要がありますが、残留許容レベルは0.03ppm未満など、非常に厳しいレベルであり、分析法もその要求を満たす高感度分析が求められます。
今回紹介した試験法は、5種類のニトロソアミン類について0.025ng/mLの定量限界を達成しました。また、データインテグリティの観点においてもGMPの要求事項を満たすデータ取得が可能となり、本手法を活用することで、厳格化の流れにあるニトロソアミンの規制に十分に対応することができると考えています。
参考情報
1)2019年9月19日 欧州医薬品庁(EMA)発
EMA/189634/2019 Notification – Request to evaluate the risk of the presence of nitrosamines in human medicinal products containing chemically synthesised APIs
2) 2021年4月23日 欧州医薬品庁(EMA)発
Questions and answers for marketing authorization holders/applicants on the CHMP Opinion for the Article 5(3) of Regulation (EC) No 726/2004 referral on nitrosamine impurities in human medicinal products
シオノギファーマは、お客様から信頼される 「技術開発型モノづくり企業(CDMO*1)」 となることをミッションとして掲げ、2019年4月1日より事業を開始しました。原薬の製造法開発および製剤処方開発から商用生産に加え、分析法開発や医薬エンジニアリング技術による設備設計サポートなどを含めた 「フルレンジサービス」 をワンストップでご提供できる体制を整えておりますので、お気軽にご相談ください。
*1 CDMO:Contract Development Manufacturing Organization






